Окулофарингеальная мышечная дистрофия (ОФМД) - заболевание, которое в мире встречается в двух генетических вариантах: аутосомно-рецессивном и аутосомно-доминантном. Эти варианты болезни являются аллельными и обусловлены различными мутациями в одном гене. Клинические варианты описаны в зависимости от типа наследования. Несмотря на то что, согласно литературным данным, ОФМД относится к более поздним формам мышечных дистрофий (4 – 5 –е десятилетие), имеется клинический пример раннего дебюта ОФМД у пациента в возрасте 23 года, с установленным диагнозом в 31 год.
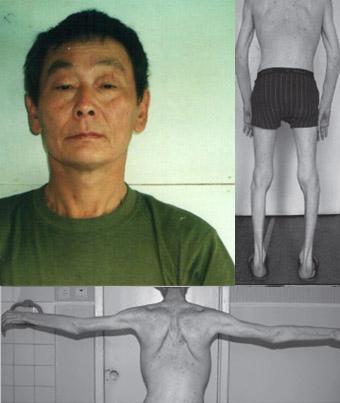 | Симптомы ОФМД: гипомимия, двусторонний птоз век, наружная офтальмоплегия, "локальная миопатия", поражающая глазодвигательные и поднимающие веки мышцы, а также мышцы-констрикторы глотки (заболевание опасно развитием нарастающей дисфагии), дисфония
При длительном течении заболевания - заметная гипотрофия нескольких групп мышц: лица, плечевого пояса, в т.ч. спины, конечностей. Часто появляются бронхо-легочные осложнения. Средний возраст манифестации: 49 ± 1,42 лет |
Особенность окулофарингеальной миодистрофии - наличие в ядрах нитевидных трубочек диаметром 8,5 нм. Аутосомно-доминантный и аутосомно-рецессивный варианты болезни обусловлены мутациями в одном и том же гене - РАВР2 (полиаденилсвязывающем протеине-2; OMIM 602 279), локализованном в области 14q11.2-q13. Основной тип мутаций - короткая экспансия тринуклеотидного повтора GCG в кодирующей части гена. В норме число повторов не превышает 6, однако у 2 % здоровых людей число повторов может достигать 7, что расценивается как проявление нормального полиморфизма. У больных с окулофарингеальной миопатией число повторов увеличено до 8–13.
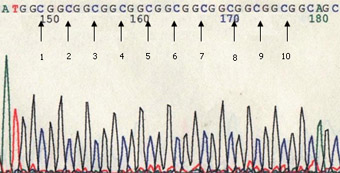


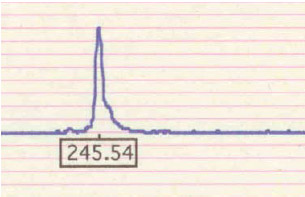
Тяжесть проявления заболевания зависит от количества повторов. Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка. Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения. На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
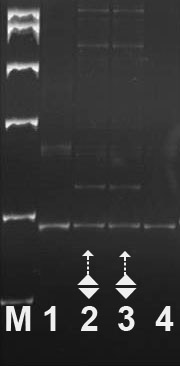 | ДНК-диагностика ОФМД в 8% ПААГ: (М - маркер PUC19/MspI) 1, 4 - генотип 6/6 GCG-повторов (здоровые). 2, 3 - генотип 6/10 GCG-повторов (больные). |
Опасность окулофарингеальной миодистрофии состоит в прогрессировании с нарастающей дисфагией и требует применения методов паллиативной неврологии. Паллиативная неврология предусматривает зондовое питание или наложение стомы. Эффективного лечения на данный момент нет. Описаны методики рассечения перстнеглоточной мышцы для улучшения глотания, но не предотвращения аспирации. Если птоз мешает зрению, используют специальные скотчевые наклейки на веки, проволочные держатели век, которые крепятся к оправе очков, либо, если нет выраженной слабости мимических мышц, прибегают к хирургическому лечению.
Аутосомно-доминантный вариант заболевания (впервыеописан Victor и соавтор. в 1962 году у 9 членов одной семьи из трех поколений ). Первые симптомы возникают на 4–5-м десятилетиях жизни и в большинстве случаев характеризуются сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечается распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Также писана (Satoyoshi & Kinoshita, 1977) семья с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. Описаны единичные больные с наличием пигментной дегенерации сетчатки. Большинство авторов эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Аутосомно-рецессивный вариант заболевания (впервые описан Fried и соавт. в 1975 году у двух сестер, родившихся от кровнородственного брака). Для этой формы болезни характерно более раннее начало и вовлечение в процесс дистальных групп мышц конечностей. При (ЭМГ) электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения. При морфологическом исследовании выявляют нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспецифичные остатки лизосомного происхождения.
Распространенность ОФМД у якутов в 10 раз выше распространенности описанной в мире. ОФМД получила накопление на территории улусов центральной Якутии и вилюйских улусов, особенно в Усть-Алданском улусе и городе Якутске.Выявлены мажорные мутации, характерные для якутской популяции: экспансия (GCG)10 в гене PÁBPN1 для окулофарингеальной миодистрофии. Распространенность аутосомно-доминантной окулофарингеальной миодистрофии в PC (Я) среди якутов составляет 11,1 на 100 тыс. населения с преимущественным накоплением в центральной и вилюйской группах улусов Якутии. Единственная молекулярно-генетическая причина всех случаев ОФМД в якутской популяции - мутация (GCG)m в 1 экзоне гена PABPN1. Выявленный единственный гаплотип является гаплотипом основателя.
Литература по ОФМД в якутской популяции:
 Russian
Russian English
English Chinese
Chinese